Integrative Genomic/Sensor Analysis (MPAI-GSA)
1 Introduction
Moving Picture, Audio and Data Coding by Artificial Intelligence (MPAI) is an international association with the mission to develop AI-enabled data coding standards. AI technologies have shown that data coding with AI-based technologies is more efficient than with existing technologies.
The MPAI approach to AI data coding standards is by defining Processing Modules called AI Modules (AIM) with standard interfaces that are combined and executed within an MPAI-specified AI-Framework. With its standards, MPAI intends to promote the development of horizontal markets of competing proprietary solutions tapping from and further promoting AI innovation.
This document describes the current plan to develop “Integrative Genomic/Sensor Analysis” (MPAI-GSA), an MPAI area of work that uses AI to understand and compress the results of data-rich experiments combining genomic/proteomic and other data, e.g. from video, motion, location, weather, medical sensors.
Chapter 2 explains the MPAI-GSA features, Chapter 3 provides summary information on the advanced IT environment that will execute MPAI-GSA applications and Chapter 4 identifies the items that will likely be the object of the MPAI-GSA standard.
2 MPAI-GSA features
Integrative Genomic/Sensor Analysis uses AI to understand and compress the results of high-throughput experiments combining genomic/proteomic and other data – for instance from video, motion, location, weather, medical sensors.
The framework consists of an API providing access to data and a protocol to specify a computation (or application) based on the data. Data can be:
- Primary, i.e. the original unprocessed high-throughput content (such as sequencing or video data)
- Secondary, i.e. the results of the pre-processing of primary data (such as gene expression estimates or features extracted from video) – applications will typically use these as input rather than primary data
- Metadata specifying additional information about the biological sample or experiment (such as sample content, cell types and barcodes, collection time and place).
The API provides uniform access to data; in particular, it standardises the definition of the semantics of the different data sources.
So far, the following application areas, ranging from personalised medicine to smart farming, have been considered:
- Integrative analysis of ‘omics datasets. It consists of complex experimental protocols combining different sources of genomic/proteomic information. One example are applications relevant to modern personalised medicine, such as determining the list and significance of the small variants present in an individual’s genome.
- Correlating high-throughput biological data with phenotypic or spatial data. It consists of applications whereby genomic or proteomic data is combined with information on the source of the biological sample (such as cell lineage for single-cell RNA-sequencing or sample content for spatial metabolomics).
- Experiments correlating genomic data with microscopic or macroscopic behaviour. It consists of protocols whereby sensor/video/MRI data is used to automatically monitor properties of lab animals (such as their macroscopic behaviour, or the functional/dynamic workings of their neural networks) and correlated with the animal’s genotype.
- Smart farming. It consists of applications combining genomic and sensor data (monitoring features such as plant/livestock phenotype or growth) in order to optimise farming yield and management.
3 AI Framework
Most MPAI applications considered so far can be implemented as a set of AIMs – AI/ML and even traditional data processing based units with standard interfaces assembled in suitable topologies to achieve the specific goal of an application and executed in an MPAI-defined AI Framework. MPAI is making all efforts to identify processing modules that are re-usable and upgradable without necessarily changing the inside logic.
MPAI plans on completing the development of a 1st generation AI Framework called MPAI-AIF in July 2021.
The MPAI-AIF Architecture is given by Figure 1
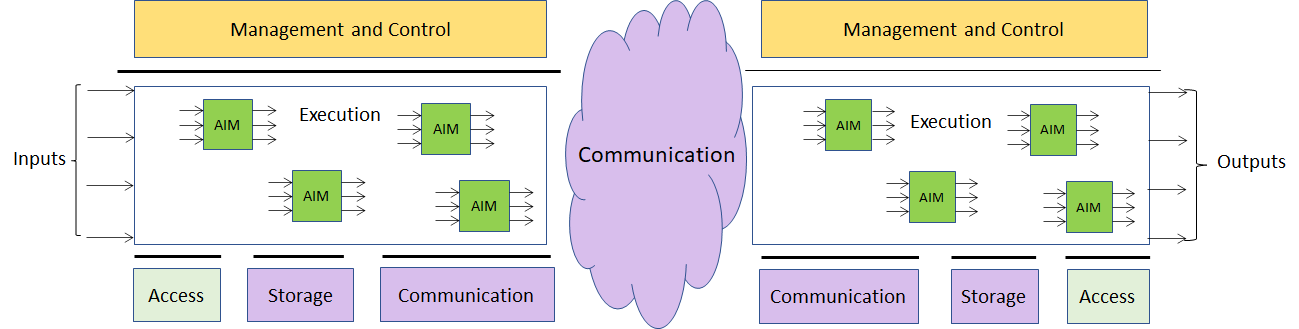
Figure 1 –The MPAI-AIF Architecture
Where
- Management and Control manages and controls the AIMs, so that they execute in the correct order and at the time when they are needed.
- Execution is the environment in which combinations of AIMs operate. It receives external inputs and produces the requested outputs both of which are application specific interfacing with Management and Control and with Communication, Storage and Access.
- AI Modules (AIM) are the basic processing elements receiving processing specific inputs and producing processing specific
- Communication is required in several cases and can be implemented, e.g. by means of a service bus and may be used to connect with remote parts of the framework
- Storage encompasses traditional storage and is used to e.g. store the inputs and outputs of the individual AIMs, data from the AIM’s state and intermediary results, shared data among AIMs.
- Access represents the access to static or slowly changing data that are required by the application such as domain knowledge data, data models, etc.
4 MPAI-GSA work plan
In this chapter we detail the four application areas. A list of AI Modules (AIMs) required across the different areas is also identified, and a first level of definition of the interfaces provided. Given that – unlike other MPAI standards – we are defining a framework where to implement applications rather than a list of applications, specifying the interfaces and a way to implement computations is sufficient to get to a full specification of the standard – the list of AIMs is only informative.
Notably, in the next sections we follow the categorisation of input data (primary, secondary and meta-) explained above. In particular, we separate primary modules, for which only data access is provided, from secondary modules – the latter implement full API and computational access.
4.1 Main areas of application
4.1.1 Integrative analysis of ‘omics datasets
In one possible realisation of this use case, one would like to correlate a list of genomic variants present in humans and having a known effect on health (metadata) with the variants present in a specific individual (secondary data). Such variants are derived from sequencing data for the individual (primary data) on which some variant calling workflow has been applied. Additional information derived from transcriptomics (RNA-sequencing, secondary data) might be taken into account. The list of variants could potentially be used to get to a personalised therapy.
Notably, there is an increasing number of companies doing just that as their core business. Their products differ by: the choice of the primary processing workflow (how to call variants from the sequencing data for the individual); the choice of the machine learning analysis (how to establish the clinical importance of the variants found); and the choice of metadata (which databases of variants with known clinical effect to use).
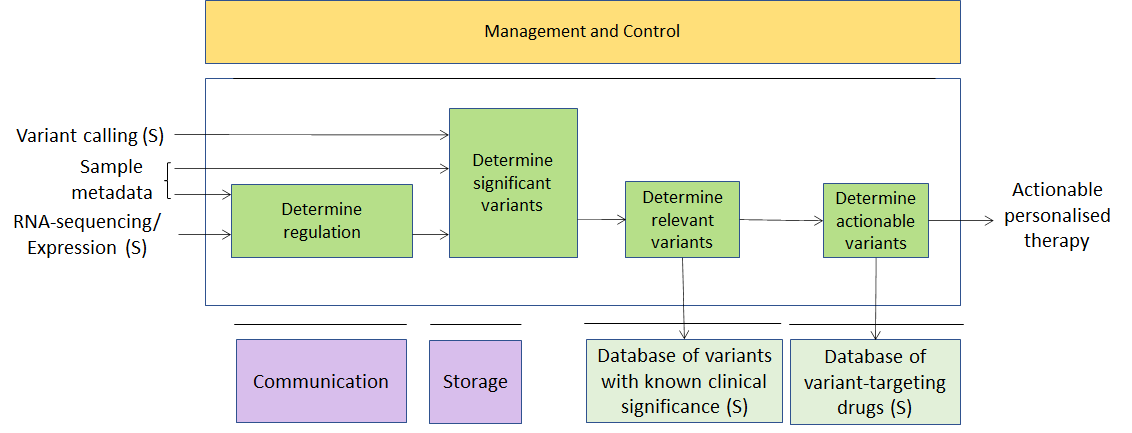
Figure 2 – A usage example of Integrative analysis of ‘omics datasets
4.1.2 Genomics and phenotypic/spatial data
As an example we take single-cell RNA sequencing. The primary data sources is RNA-sequencing performed at the same time on a number (typically hundred of thousands) of different cells – while bulk RNA sequencing mixes together RNAs coming from several thousands of different cells, in single-cell RNA sequencing the RNAs coming from each different cell are separately barcoded, and hence distinguishable. The DNA barcodes for each cell would be metadata here. Cells can then be clustered together according to the expression patterns present in the secondary data (vectors of expression values for all the species of RNA present in the cell) and, if sufficient metadata and spatial information is present, clusters of expression patterns can be associated with different types/lineages of cells – the technique is typically used to study tissue differentiation. A number of complex algorithms exist to perform primary analysis (statistical uncertainty in single-cell RNA-sequencing is much bigger than in bulk RNA-sequencing) and, in particular, secondary AI-based clustering/analysis. Again, expressing those algorithms in terms of MPAI-GSA would make them much easier to describe and much more comparable. External commercial providers might provide researchers with clever modules to do all or part of the machine learning analysis.
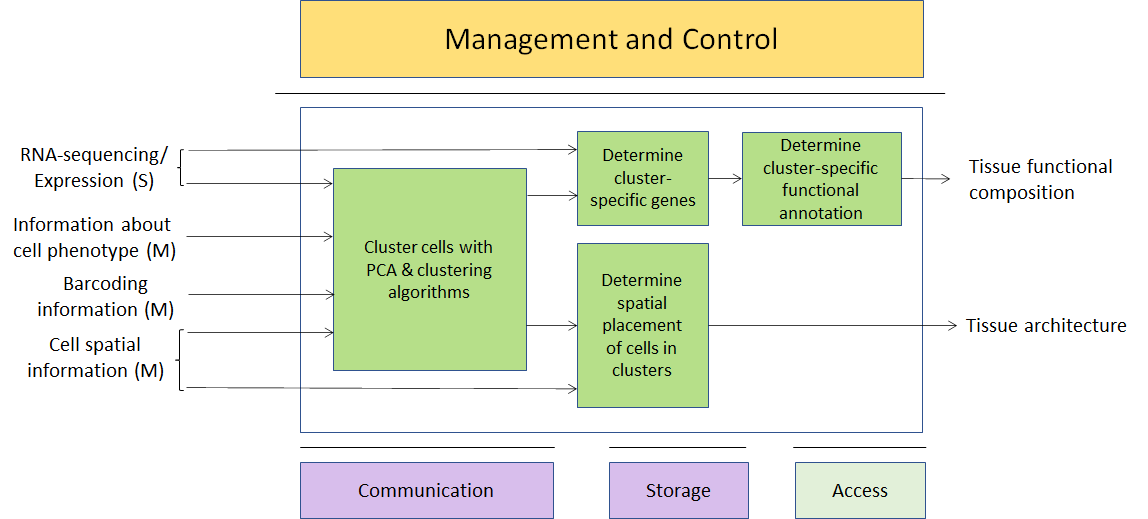
Figure 3 – A usage example of Genomics and Phenotypic/spatial data
4.1.3 Genomics and behaviour
In a typical application of this use case, one would like to correlate animal behaviour (typically of lab mice) with their genetic profile (case of knock-down mice). Another application might be correlating genetic variants with the reaction to drug administration (typically encountered in neurobiology), possibly monitored in real-time with functional MRI scans. Hence primary data would be video data from cameras tracking the animal and/or data from an MRI scanner; secondary data would be processed video data in the form of primitives describing the animal’s movement, well-being, activity, weight, etc.; and metadata would be a description of the genetic background of the animal (for instance, the name of the gene which has been deactivated) or a timeline with the list and amount of drugs which have been administered to the animal. Again, there are several companies providing software tools to perform some or all of such analysis tasks – they might be easily reformulated in terms of MPAI-GSA applications.
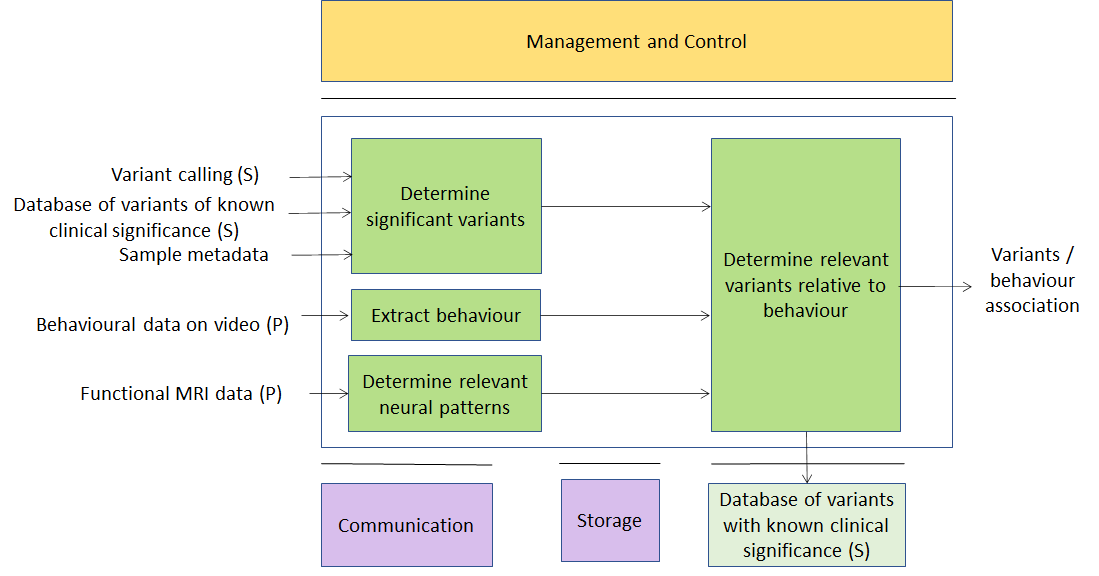
Figure 4 – A usage example of Genomics and Behaviour
4.1.4 Smart Farming
During the past few years, there has been an increasing interest in data-rich techniques to optimise livestock and crop production (so called “smart farming”). The range of techniques is constantly expanding, but the main ideas are to combine molecular techniques (mainly high-throughput sequencing and derived protocols, such as RNA-sequencing, ChIP-sequencing, HiC, etc.; and mass-spectrometry – as per the ‘omics case at point 2) and monitoring by images (growth rate under different conditions, sensor data, satellite-based imaging) for both livestock species and crops. So this use case can be seen as a combination of cases 2 and 4. Primary sources would be genomic data and images; secondary data would be vectors of values for a number of genomic tags and features (growth rate, weight, height) extracted from images; metadata would be information about environmental conditions, spatial position, etc. A growing number of companies are offering services in this area – again, having the possibility of deploying them as MPAI-GSA applications would open up a large arena where academic or commercial providers would be able to meet the needs of a number of customers in a well-defined way.
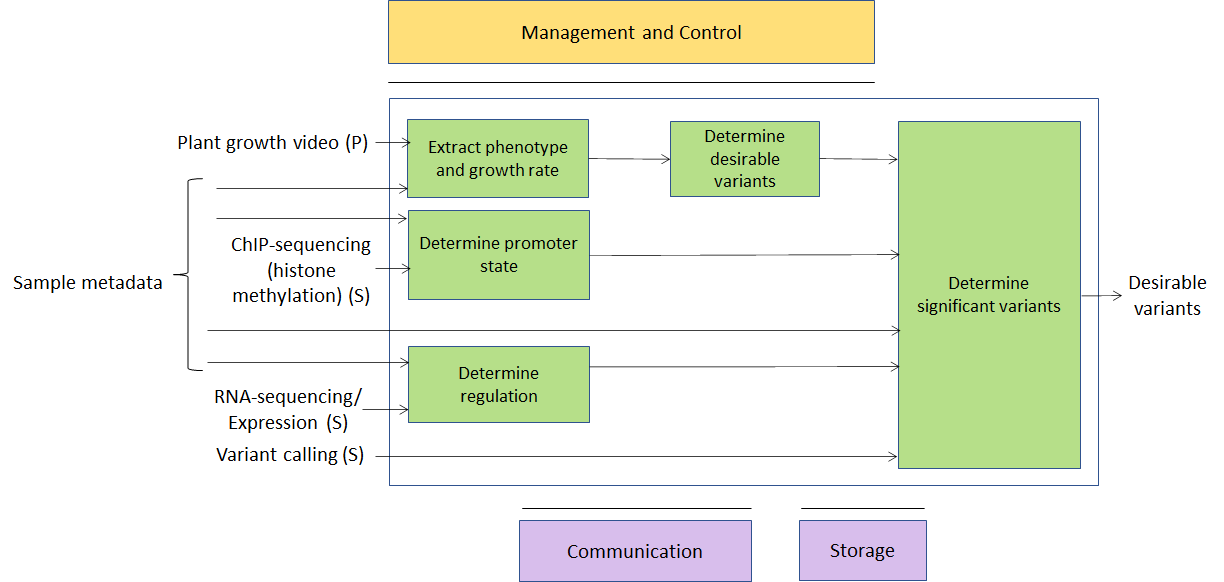
Figure 4 – A usage example of Smart Farming
4.2 Definition of AIMs in terms of simpler AIMs
The modules presented in the previous section are very high-level, and typically, each one of them might correspond to complex analysis methods implemented in terms of a number of simpler AIMs. For instance, in a real-life scenario the block “Determine significant variants” in Figure 3 would correspond to a complex “pipeline”, in bioinformatics jargon, involving the use of a number of algorithms and programs. In addition, several methods to implement the same block would typically exist and be accepted in the literature.
MPAI-GSA does not really concern itself with the implementation of each module – it only defines:
- The possible categories of data sources (be they primary, secondary or metadata) and their semantics
- A way to specify and run AIMs in terms of data sources and computational methods operating on them. AIMs can also be combined into more complex AIMs thanks to the same API.
While ideally (2) is done by taking advantage of the functionality offered by MPAI-AIF, (1) requires a number of external data formats and sources to be described and understood by the MPAI-GSA. This is done in the next section.
4.3 Main low-level use cases and their input/output data categories
In this section, we offer a collection of input/output data categories that are likely to be needed in order to support the high-level use cases presented so far. We enumerate them based on a list of low-level use cases, corresponding to more basic AIMs that would be relevant to each areas of application of MPAI-GSA.
Consistent with the general structure of MPAI-GSA, we follow the categorisation of input data as primary, secondary, and metadata. In particular, we separate primary modules, for which only data access is provided, from secondary modules – the latter implement full API and computational access.
4.3.1 K-mer based analysis
4.3.1.1 Compute k-mer frequency (P)
| Function | Derive the distribution of k-mer frequencies from sequencing reads |
| Primary inputs | FASTA/FASTQ (reads) |
| Primary outputs | CSV (list of k-mer, frequency) |
| Notes | Only access and metadata supported |
4.3.2 Genome assembly and annotation
4.3.2.1 De-novo assembly (P)
| Function | Derive a new reference for the species/individual by assembling sequencing reads |
| Primary inputs | FASTA/FASTQ (reads) |
| Primary outputs | FASTA (assembly), graph formats (assembly) |
| Notes | Only access and metadata supported |
4.3.2.2 De-novo annotation (P)
| Function | Derive a genomic annotation for a newly assembled genome |
| Primary inputs | FASTA (reads, reference), GFF/GTF3 (genome annotation) |
| Primary outputs | GFF/GTF3 (genome reannotation) |
| Notes | Only access and metadata supported |
4.3.3 Genome re-sequencing
4.3.3.1 Variant calling (P)
| Function | Determine (“call”) genomic variants for an individual (i.e. differences between the reference genome for the species and the genome of an individual) |
| Primary inputs | FASTQ (reads), FASTA (reference) |
| Primary outputs | VCF (deduced variants) |
| Notes | Only access and metadata supported |
4.3.3.2 (Single-cell) RNA-sequencing, expression (P)
| Function | Derive a list of expression values for all (reannotated) genes/isoforms for each condition |
| Primary inputs | FASTQ (reads), CSV (metadata), FASTA (reference), GFF/GTF3 (genome annotation) |
| Primary outputs | CSV (expression), BigWig (tracks) |
| Notes | Only access and metadata supported |
4.3.3.3 Single-cell RNA-sequencing, clustering (S)
| Function | Derive a clustering for the cells studied during the experiment (possibly informed by position) |
| Secondary inputs | CSV (expression, high-dimensional plots) |
| Secondary outputs | CSV (cell clustering) |
4.3.3.4 BS-sequencing (P)
| Function | Derive a signal (“track”) describing the level of methylation along the genome |
| Primary inputs | FASTQ (reads), CSV (metadata), FASTA (reference), GFF/GTF3 (genome annotation) |
| Primary outputs | BigWig (tracks) |
| Notes | Only access and metadata supported |
4.3.3.5 ChIP-sequencing (P)
| Function | Derive a signal (“track”) describing the level of interaction between the target protein and DNA along the genome |
| Primary inputs | FASTQ (reads), CSV (metadata), FASTA (reference), GFF/GTF3 (genome annotation) |
| Primary outputs | BigWig (tracks) |
| Notes | Only access and metadata supported |
4.3.3.6 HiC, contact matrices (P)
| Function | Derive information on spatial connections between different regions of the genome |
| Primary inputs | FASTQ (reads), CSV (metadata), FASTA (reference), GFF/GTF3 (genome annotation) |
| Primary outputs | Matrix formats such as MatrixMarket (position-to-position links) |
| Notes | Only access and metadata supported |
4.3.4 Personalised genomics
4.3.4.1 Determine variant significance (S)
| Function | Correlate individual variants with databases of variants with known clinical significance |
| Secondary inputs | VCF (known variants), VCF (deduced variants) |
| Secondary outputs | CSV (list of significant variants, clinical significance) |
4.3.5 Integrative analysis
4.3.5.1 Determine differential expression/signals (S)
| Function | Determine differential signals in RNA-, ChIP-, BS-sequencing experiments, cluster genes/samples accordingly |
| Secondary inputs | Corresponding primary outputs (expression values as CSV, genome tracks as BigWig) |
| Secondary outputs | CSV |
4.3.5.2 Perform pathway/enrichment/network analysis (S)
| Function | Determine clusters/pathways of enriched genes, and their functional connection |
| Secondary inputs | Corresponding primary outputs ([SC] RNA-sequencing) |
| Secondary outputs | CSV, graph formats |
4.3.5.3 Combine different primary sources (S)
| Function | Combine signal tracks or expression values for the same sample coming from different sequencing protocols; cluster genes/samples accordingly |
| Secondary inputs | Corresponding primary inputs (expression values as CSV, genome tracks as BigWig) |
| Secondary outputs | BigWig, CSV |
4.3.5.4 Study time series (S)
| Function | Combine signal tracks or expression values for the same biological system coming from different time points; cluster genes/samples accordingly |
| Secondary inputs | Corresponding primary inputs (expression values as CSV, genome tracks as BigWig) |
| Secondary outputs | BigWig, CSV |
4.3.6 Automatic analysis of animal behaviour
4.3.6.1 Animal dynamics
| Function | To detect the animal and its spatial motion within the observation field, possibly within a specified ROI |
| Primary inputs | Video signal as stream or file, ROI |
| Primary outputs/Secondary inputs | Distance, (average) velocity, acceleration, time spent, time spent near walls, trajectories, turning speed (everywhere and/or in ROI) |
4.3.6.2 Area and perimeter
| Function | To detect areas where the animal preferentially dwells during the observed time |
| Primary inputs | Video signal as stream or file |
| Primary outputs/Secondary inputs | Coordinates, area and perimeter |
4.3.6.3 ID Tracker
| Function | To detect and track a specific animal, alone or among many (unsupervised or based on tracking devices) |
| Primary inputs | Video signal as stream or file |
| Primary outputs/Secondary inputs | Identification of animal (everywhere and/or in ROI) |
4.3.6.4 Behaviour detection
| Function | To analyse and detect the behaviour of one specified, or more, of the animals present within the observation field |
| Primary inputs | Video signal as stream or file |
| Primary outputs/Secondary inputs | Bites, persecution, sexual behaviour, angle of turn, grooming, jump, walk, immobilization, and touch |
4.4 Summary of input/output data categories
The data categories identified in the last section can be summarised in the next table.
| Data Type | Used to represent |
| FASTA | Sequencing reads; Genomic references; Genomic assemblies |
| FASTQ | Sequencing reads |
| GFF/GTF | Genomic functional annotations |
| VCF | Genomic variants |
| BigWig | Genomic tracks |
| Graph formats | Genomic assemblies |
| (Sparse) MATRIX FORMATS | Genomic contacts; Expression values |
| CSV/tabular FORMATS | Location/satellite data; Sensor data; Metadata; Expression values; Clustering results; List of audio/video events; Time series; Sets (cells; pathways; conditions) |
| AUDIO/VIDEO FORMATS | Experiment recording |
| MRI-like formats | Experiment recording |
| SUBTITLE-LIKE formats | Association between audio/video events and audio/video streams |
5 Conclusions
The document in its current form is work in progress. MPAI intends to add more details to the existing and to add more usage examples to be covered by the future MPAI-GSA standard.
When the document will be considered sufficiently mature, MPAI will issue a Call for Technologies requesting MPAI members and the industry to submit proposals for:
- Data formats suitable as inputs and outputs of the identified AIMs
- Additions or removal of input/output signals to the identified AIMs with identification of data formats required by the new input/output signals
- Possible alternative partitioning of the AIMs implementing the example cases providing
- Arguments in support of the proposed partitioning
- Detailed specifications of the inputs and outputs of the proposed AIMs
- New Use Cases fully described as in the final version of this document.
Respondents will be asked to state in their submissions their intention to adhere to the Framework Licence developed for MPAI-GSA when licencing their technologies if included in the MPAI-GSA standard. Please note that “a Framework Licence is the set of conditions of use of a licence without the values, e.g. currency, percent, dates etc.”. The Framework Licence will give the MPAI-GSA standard a clear IPR licensing framework.
The MPAI-GSA Framework Licence will be developed, as for all other MPAI Framework Licences, in compliance with the generally accepted principles of competition law.