Integrative genomic/sensor analysis (MPAI-GSA)
Proponent: Paolo Ribeca (BioSS/James Hutton)
Description: Most experiment in quantitative genomics consist of a setup whereby a small amount of metadata – observable clinical score or outcome, desirable traits, observed behaviour – is correlated with, or modelled from, a set of data-rich sources. Such sources can be:
- Biological experiments – typically sequencing or proteomics/metabolomics data
- Sensor data – coming from images, movement trackers, etc.
All these data-rich sources share the following properties:
- They produce very large amounts of “primary” data as output
- They need “primary”, experiment-dependent, analysis, in order to project the primary data (1) onto a single point in a “secondary”, processed space with a high dimensionality – typically a vector of thousands of values
- The resulting vectors, one for each experiment, are then fed to some machine or statistical learning framework, which correlates such high-dimensional data with the low-dimensional metadata available for the experiment. The typical purpose is to either model the high-dimensional data in order to produce a mechanistic explanation for the metadata, or to produce a predictor for the metadata out of the high-dimensional data.
- Although that is not typically necessary, in some circumstances it might be useful for the statistical or machine learning algorithm to be able to go back to the primary data (1), in order to extract more detailed information than what is available as a summary in the processed high-dimensional vectors produced in (2).
Providing a uniform framework to:
- Represent the results of such complex, data-rich, experiments, and
- Specify the way the input data is processed by the statistical or machine learning stage
would be extremely beneficial.
Comments: Although this structure above is common to a number of experimental setups, it is conceptual and never made explicit. Each “primary” data source can consist of heterogeneous information represented in a variety of formats, especially when genomics experiments are considered, and the same source of information is usually represented in different ways depending on the analysis stage – primary or secondary. That results in data processing workflows that are ad-hoc – two experiments combining different sets of sources will require two different workflows able to process each one a specific combination of input/output formats. Typically, such workflows will also be layered out as a sequence of completely separated stages of analysis, which makes it very difficult for the machine or statistical learning stage to go back to primary data when that would be necessary.
MPAI-GSA aims to create an explicit, general and reusable framework to express as many different types of complex integrative experiments as possible. That would provide (I) a compressed, optimized and space-efficient way of storing large integrative experiments, but also (II) the possibility of specifying the AI-based analysis of such data (and, possibly, primary analysis too) in terms of a sequence of pre-defined standardized algorithms. Such computational blocks might be partly general and prior-art (such as standard statistical algorithms to perform dimensional reduction) and partly novel and problem-oriented, possibly provided by commercial partners. That would create an healthy arena whereby free and commercial methods could be combined in a number of application-specific “processing apps”, thus generating a market and fostering innovation. A large number of actors would ultimately benefit from the MPAI-GSA standard – researchers performing complex experiments, companies providing medical and commercial services based on data-rich quantitative technologies, and the final users who would use instances of the computational framework as deployed “apps”.
Examples
The following examples describe typical uses of the MPAI-GSA framework.
- Medical genomics – sequencing and variant-calling workflows
In this use case, one would like to correlate a list of genomic variants present in humans and having a known effect on health (metadata) with the variants present in a specific individual (secondary data). Such variants are derived from sequencing data for the individual (primary data) on which some variant calling workflow has been applied. Notably, there is an increasing number of companies doing just that as their core business. Their products differ by: the choice of the primary processing workflow (how to call variants from the sequencing data for the individual); the choice of the machine learning analysis (how to establish the clinical importance of the variants found); and the choice of metadata (which databases of variants with known clinical effect to use). It would be easy to re-deploy their workflows as MPAI-GSA applications.
- Integrative analysis of ‘omics datasets
In this use case, one would like to correlate some macroscopic variable observed during a biological process (e.g. the reaction to a drug or a vaccine – metadata) with changes in tens of thousands of cell markers (gene expression estimated from RNA; amount of proteins present in the cell – secondary data) measured through a combination of different high-throughput quantitative biological experiments (primary data – for instance, RNA-sequencing, ChIP-sequencing, mass spectrometry). This is a typical application in research environments (medical, veterinary and agricultural). Both primary and secondary analysis are performed with a variety of methods depending on the institution and the provider of bioinformatics services. Reformulating such methods in terms of MPAI-GSA would help reproducibility and standardisation immensely. It would also provide researchers with a compact way to store their heterogeneous data.
- Single-cell RNA-sequencing
Similar to the previous one, but in this case at least one of the primary data sources is RNA-sequencing performed at the same time on a number (typically hundred of thousands) of different cells – while bulk RNA sequencing mixes together RNAs coming from several thousands of different cells, in single-cell RNA sequencing the RNAs coming from each different cell are separately barcoded, and hence distinguishable. The DNA barcodes for each cell would be metadata here. Cells can then be clustered together according to the expression patterns present in the secondary data (vectors of expression values for all the species of RNA present in the cell) and, if sufficient metadata is present, clusters of expression patterns can be associated with different types/lineages of cells – the technique is typically used to study tissue differentiation. A number of complex algorithms exist to perform primary analysis (statistical uncertainty in single-cell RNA-sequencing is much bigger than in bulk RNA-sequencing) and, in particular, secondary AI-based clustering/analysis. Again, expressing those algorithms in terms of MPAI-GSA would make them much easier to describe and much more comparable. External commercial providers might provide researchers with clever modules to do all or part of the machine learning analysis.
- Experiments correlating genomics with animal behaviour
In this use case, one wants to correlate animal behaviour (typically of lab mice) with their genetic profile (case of knock-down mice) or the previous administration of drugs (typically encountered in neurobiology). Hence primary data would be video data from cameras tracking the animal; secondary data would be processed video data in the form of primitives describing the animal’s movement, well-being, activity, weight, etc.; and metadata would be a description of the genetic background of the animal (for instance, the name of the gene which has been deactivated) or a timeline with the list and amount of drugs which have been administered to the animal. Again, there are several companies providing software tools to perform some or all of such analysis tasks – they might be easily reformulated in terms of MPAI-GSA applications.
- Spatial metabolomics
One of the most data-intensive biological protocols nowadays is spatial proteomics, whereby in-situ mass-spec/metabolomics techniques are applied to “pixels”/”voxels” of a 2D/3D biological sample in order to obtain proteomics data at different locations in the sample, typically with sub-cellular resolution. This information can also be correlated with pictures/tomograms of the sample, to obtain phenotypical information about the nature of the pixel/voxel. The combined results are typically analysed with AI-based technique. So primary data would be unprocessed metabolomics data and images, secondary data would be processed metabolomics data and cellular features extracted from the images, and metadata would be information about the sample (source, original placement within the body, etc.). Currently the processing of spatial metabolomics data is done through complex pipelines, typically in the cloud – having these as MPAI-GSA applications would be beneficial to both the researchers and potential providers of computing services.
- Smart farming
During the past few years, there has been an increasing interest in data-rich techniques to optimise livestock and crop production (so called “smart farming”). The range of techniques is constantly expanding, but the main ideas are to combine molecular techniques (mainly high-throughput sequencing and derived protocols, such as RNA-sequencing, ChIP-sequencing, HiC, etc.; and mass-spectrometry – as per the ‘omics case at point 2) and monitoring by images (growth rate under different conditions, sensor data, satellite-based imaging) for both livestock species and crops. So this use case can be seen as a combination of cases 2 and 4. Primary sources would be genomic data and images; secondary data would be vectors of values for a number of genomic tags and features (growth rate, weight, height) extracted from images; metadata would be information about environmental conditions, spatial position, etc. A growing number of companies are offering services in this area – again, having the possibility of deploying them as MPAI-GSA applications would open up a large arena where academic or commercial providers would be able to meet the needs of a number of customers in a well-defined way.
Requirements:
MPAI-GSA should provide support for the storage of, and access to:
- Unprocessed genomic data from the most common sources (reference sequences, short and long sequencing reads)
- Processed genomic data in the form of annotations (genomic models, variants, signal tracks, expression data). Such annotations can be produced as the result of primary analysis of the unprocessed data or come from external sources
- Video data both unprocessed and processed (extracted features, location, movement tracking)
- Sensor data both unprocessed (such as GPS position tracking, series describing temperature/humidity/weather conditions, general input of multi-channel sensors) and processed.
- Experiment meta-data (such as collection date and place; classification in terms of a number of user-selected categories for which a discrete or continuous value is available)
- Support for the semantic description of the ontology of all the considered sources.
MPAI-GSA should also provide support for:
- The combination into a general analysis workflow of a number of computational blocks that access processed, and possibly unprocessed, data as input channels, and produce output as a sequence of vectors in a space of arbitrary dimension. Combination would be done in terms of nodes (processing blocks and adaptors [blocks that return as output a subset of the input channels, nodes that replicate the input as output several times]), and a connection graph
- The possibility of defining and implementing a novel processing block from scratch in terms of either some source code or a proprietary binary codec
- A number of pre-defined blocks that implement well-known analysis methods (such as PCA, MDS, CA, SVD, NN-based methods, etc.).
Object of standard: A high-level, schematic description of the standard can be found in the following figure:
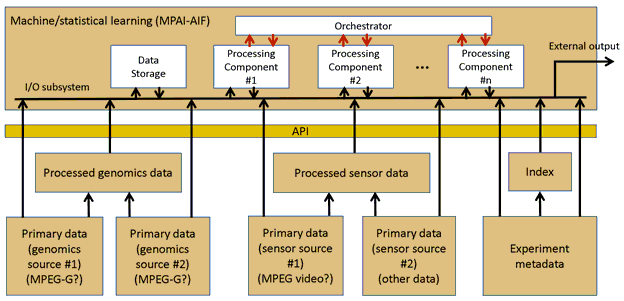
Figure 1 – A reference diagram for MPAI-GSA
Currently, three areas of standardization are identified:
- Interface to define the machine/statistical learning in terms of basic algorithmic blocks:
- Ability to define basic computational blocks that take a (sub)set of inputs and produce a corresponding set of outputs (the cardinalities of the sets can be different). More in detail:
- The way of defining a block will be mandated by the standard (in particular, a unique ID might be issued to the implementer by a centralised authority)
- A basic computational block will need to define and export its computational requirements in terms of CPU and RAM
- Some blocks might specify their preferred execution model, for instance some specific cloud computation platform
- Some pre-defined blocks might be provided by the standard, for instance the implementation of a few well-known methods to perform dimensional reduction (PCA, MDS, etc.).
- Ability to define basic computational blocks that take a (sub)set of inputs and produce a corresponding set of outputs (the cardinalities of the sets can be different). More in detail:
- Ability to create processing pipelines in terms of basic algorithmic blocks and to define the associated control flow required to perform the full computation
- A standardised output format
- Whenever possible, interoperability with established technologies (e.g. TensorFlow).
The components for this area will very likely be provided by MPAI-AIF. MPAI-AIF proposes a general, standardised framework that can be used to specify computational workflows based on machine learning in a number of scenarios. It will provide core functionality to several of the MPAI standard proposals currently under consideration.
- Interface for the machine/statistical learning to access processed (secondary) data: A first set of input and output signals, with corresponding syntax and semantics, to process secondary data (i.e., the results of processing primary data) with methods based on machine learning. They have the following shared properties:
- Irrespective of their source (genomic or sensor) all the inputs to the AI processor are expressed as vectors of values of different number types
- Information about the semantics of each input (which source produced which input, and which biological entity/feature each input corresponds to) is standardised and provided through metadata
- In order to provide fast access and search capabilities, an index for the metadata is provided.
- Interface for the machine/statistical learning to access unprocessed (primary) data with the following features:
- Definition of the semantics of the primary data accessible for a large number of data types (especially genomic – see ISO/IEC 23092 part 6 for an indicative list)
- Information on the methods used to process the primary data into secondary with a clearly defined semantics is standardised and provided through metadata
- Possibly, ability to define basic computational blocks for primary analysis similar to those at (2) and their combination into complex computations, in order to re-process primary data whenever needed by (2)
- The processing pipeline may be a combination of local and in-cloud processing.
On the other hand, we would not like to re-standardise the representation of primary data! For instance, part 6 of the the MPEG-G format (ISO/IEC 23092) already standardizes meta-data for most of the techniques employed in the field of genomics under the unifying concept of genomic annotations. Part 3 already offers a clear API through which primary sequencing data can be accessed. In order to avoid effort replication, MPAI-GSA might represent genomic data as MPEG-G encoded data. Similar possibilities might be considered for video primary sources.
Benefits: MPAI-GSA will be offer a number of advantages to a number of actors:
- Technology providers and researchers will have available a robust, tested framework within which to develop modular applications that are easy to define, deploy and modify. They will no longer need to spend time and resources on implementing access to a number of basic data formats or the mechanics of a unified access to heterogeneous resources – they will be able to focus fully on the development of their machine learning methods. The methods will be clearly defined in terms of computational modules, some of which can be provided by commercial third parties. This will drastically improve reproducibility, which is an increasing problem with the current biological research based on big data. Offering a robust, well-defined framework will also lower the amount of resources needed to enter the market and help smaller actors to generate and implement competitive ideas
- Equipment manufacturers and application vendors can choose from the set of technologies made available through MPAI-GSA standard by different sources, integrate them and satisfy their specific needs
- Service providers will have available a growing number of MPAI-GSA applications able to solve different categories of data analysis problems. As all such applications are clearly expressed in terms of a reproducible standard rather than being developed and hosted opaquely on some closed corporate computing infrastructure, comparing the different applications and offering different options to customers will become much simpler
- End users will enjoy a thriving, competitive market that provides an ever-growing number of quality solutions by an ever-growing number of actors. The general availability of such a powerful technology will hopefully make widespread applications that today require research computational equipment and personnel, such as clinically oriented genetic/genomic analysis.
Bottlenecks: In order to fully exploit the potential of MPAI-GSA, one will need widespread availability of computing power, in particular for applications comprising steps whereby primary data is processed into secondary. That would typically require the ability to perform some of such computational steps in the cloud, as few users have access to enough resources. AI-friendly processing units able to implement and speed up secondary analysis would also help, perhaps allowing MPAI-GSA applications based only on secondary-analysis on commodity devices such as mobile phones.
Social aspects: Genomic applications partially based on the phone might facilitate social uses of the technology (such as receiving and exploring the results of genetic tests, or establishing genealogies).
Success criteria: Data-rich applications are the future of a number of disciplines, in particular life sciences, personalised medicine and the one-health approach – whereby humans, livestock, farming and ultimately the whole terrestrial ecosystem is seen as an integrated system, with each part influencing the rest. At the moment, however, creating analysis workflows able to exploit the data is a painstaking ad-hoc process which requires sizeable investments in technology and development. Most of the times the effort cannot be reused, as by definition the applications are problem-specific. MPAI-GSA will be successful if it can create a framework facilitating the development of modular, reusable analysis frameworks that on one hand trivialise data storage and on the other hand streamline the creation of complex methods. Its success will be defined by its ability to attract a number of actors – researchers, commercial providers of computational solutions and analytical services, end users. The thriving ecosystem of applications thus generated will be a necessary ingredient to transparently integrate data-rich technologies for the life sciences into common practice, widespread appliances, and everyday life.